ГОСТ Р ИСО 13408-1-2000
Группа Р26
ГОСУДАРСТВЕННЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕPAЦИИ
АСЕПТИЧЕСКОЕ ПРОИЗВОДСТВО МЕДИЦИНСКОЙ ПРОДУКЦИИ
Часть 1
Общие требования
Aseptic processing of health care products.
Part 1. General requirements
ОКС 11.080
ОКП 94 5120
Дата введения 2002-01-01
Предисловие
1 ПОДГОТОВЛЕН Ассоциацией инженеров по контролю микрозагрязнений (АСИНКОМ)
ВНЕСЕН Техническим комитетом по стандартизации ТК 383 "Стерилизация медицинской продукции" Госстандарта России
2 ПРИНЯТ И ВВЕДЕН В ДЕЙСТВИЕ Постановлением Госстандарта России от 25 сентября 2000 г. N 232-ст
3 Настоящий стандарт содержит аутентичный текст международного стандарта ИСО 13408-1-98 "Асептическое производство медицинской продукции. Часть 1: Общие требования"
4 ВВЕДЕН ВПЕРВЫЕ
Введение
Введение
Медицинская продукция, на этикетке которой указано "Стерильно", должна быть изготовлена соответствующими валидированными методами. Технический комитет ИСО/ТК 198 подготовил стандарты по финишной стерилизации (стерилизации в упаковке) медицинской продукции радиацией (ГОСТ Р ИСО 11137-2000), влажным теплом (ГОСТ Р ИСО 11134-2000), жидкими химическими стерилизующими веществами (ИСО 14160-98) и оксидом этилена (ГОСТ Р ИСО 11135-2000). Если медицинская продукция должна быть стерильной, но не подлежать финишной стерилизации, используется асептическое производство. Существуют две ситуации, при которых используется асептическое производство:
a) асептическая подготовка растворов и наполнение ими;
b) асептические приготовление, перемещение и упаковка твердых продуктов, которые не подлежат финишной стерилизации в окончательной упаковке.
Асептическое производство требует предварительной стерилизации всех частей или компонентов продукции, которые находятся в прямом контакте с продуктом, упаковываемым в асептических условиях. Продукт производится в контролируемой окружающей среде, в которой загрязнение частицами и микроорганизмами не должно превышать определенных уровней и участие человека сведено до минимума.
Асептическое производство требует особой аккуратности и ответственности. В производстве используются валидированные системы, персонал, обученный соответствующим образом, контролируемые окружающие среды и полностью документированные технологические процессы.
В то время как при финишной стерилизации используются процессы с известной летальностью (эффективностью уничтожения микроорганизмов), стерильность в асептическом производстве может быть обеспечена только за счет помещений, оборудования и персонала, связанных с процессом. Следует также использовать данные о продукте, чтобы сохранить стерильность в контейнере и/или закрытой системе в соответствии с асептическими принципами.
При асептическом производстве учитывают следующие основные факторы:
a) обучение персонала;
b) планировочные решения и характеристики здания, помещений и оборудования;
c) программу контроля загрязнений частицами и микроорганизмами;
d) системы подготовки воды, пара, воздуха и других технологических газов;
e) описание технологических операций и инструкции по их выполнению, включая персонал, материалы, потоки материалов, подготовку растворов и соответствующие критерии приемки;
f) использование и валидацию процессов стерилизации, включая методы дезинфекции;
g) методы валидации и требования к имитаторам наполнения и системам контейнер/укупорка;
h) эксплуатационные инструкции по критериям приемки, исследовательские отчеты и решения по выпуску/отзыву продукта.
Стандарт ИСО 13408-1 был подготовлен Техническим комитетом ИСО/ТК 198 "Стерилизация медицинской продукции". ИСО 13408 "Асептическое производство медицинской продукции" состоит из следующих частей:
- Часть 1: Общие требования
- Часть 2: Фильтрация
- Часть 3: Лиофилизация
- Часть 4: Стерилизация и очистка на месте
- Часть 5: Асептическое производство твердых медицинских изделий
- Часть 6: Изолирующие/барьерные технологии
1 Область применения
Настоящий стандарт устанавливает общие требования к процессам, программам и процедурам валидации и контроля в асептическом производстве медицинской продукции.
Примечание - Последующие части стандарта будут посвящены специальным вопросам асептического производства, включая детализированное описание различных специфических процессов и методов, относящихся к фильтрации, лиофилизации, стерилизации и очистке на месте, изолирующей технологии и производству твердых медицинских изделий.
Настоящий стандарт не заменяет требования национальных регламентов, таких как Правила надлежащего производства - Good Manufacturing Practice (GMP) и/или руководства, которые относятся к сфере определенной национальной или региональной юрисдикции.
2 Определения
В настоящем стандарте используют следующие термины с соответствующими определениями:
2.1 уровень действия (action level) "контроль окружающей среды": Установленный уровень загрязнения микроорганизмами или частицами, при превышении которого требуется немедленное вмешательство и корректирующие действия.
2.2 уровень действия (action level) "наполнение средами": Установленный уровень или число нестерильных проб при наполнении средами (имитации наполнения), при превышении которого необходимо исследовать причину нестерильности и выполнить корректирующие действия.
2.3 уровень предупреждения (alert level) "контроль окружающей среды": Установленный уровень загрязнения микроорганизмами или частицами, который дает раннее предупреждение о потенциальной тенденции отклонения от нормальных эксплуатационных условий, при котором не требуется обязательное определенное корректирующее действие, но может потребоваться проведение исследования причин такого отклонения.
2.4 уровень предупреждения (alert level) "при работе с наполнением средами": Установленный уровень или число нестерильных проб при наполнении средами (имитации наполнения), при достижении которого необходимо установить причину нестерильности, но выполнять определенные корректирующие действия необязательно.
2.5 асептическое наполнение (aseptic filling): Часть асептического процесса, при котором предварительно стерилизованный продукт наполняется и/или он упаковывается в стерильные контейнеры и укупоривается.
2.6 линия асептического наполнения (aseptic filling line): Технологическое оборудование или установка, где в асептических условиях проводится наполнение контейнеров и/или медицинских изделий.
Примечание - Обычно линия асептического наполнения строится так, что процесс наполнения контейнеров и/или медицинских изделий проходит вдоль линии (отсюда термин "линия").
2.7 асептическое производство (aseptic processing): Асептическое наполнение контейнеров для продукта и/или упаковка изделий в контролируемой окружающей среде, в которой обеспечение воздухом, материалами, оборудованием и персоналом регулируется так, чтобы загрязнение микроорганизмами и частицами не выходило за установленные пределы.
2.8 зона асептического производства - ЗАП (aseptic processing area - АРА): Контролируемая окружающая среда для асептического производства, состоящая из нескольких зон, в которых обеспечение воздухом, материалами, оборудованием и персоналом регулируется так, чтобы загрязнение микроорганизмами и частицами не выходило за установленные пределы.
2.9 протокол изготовления серии - партии (batch manufacturing record): Производственная документация, которая свидетельствует, что серия или партия продукта изготовлена в соответствии с установленным технологическим процессом и требованиями к обеспечению качества.
2.10 бионагрузка (bioburden): Популяция живых микроорганизмов на медицинском продукте или упаковке до стерилизации.
2.11 бионагрузка (bioburden): Популяция живых микроорганизмов на материалах и оборудовании, поступивших в зону асептического производства (ЗАП).
2.12 биологический индикатор (biologocal indicator): Готовый к применению инокулированный носитель в первичной упаковке, обеспечивающий определенную резистентность (устойчивость) к конкретному режиму стерилизации.
Примечание - Микроорганизмы часто используются на носителе, который представляет собой материал подложки с находящимися на нем тест-организмами.
2.13 форма контейнера (container configuration): Конструкция контейнера независимо от емкости.
Примечание - Поскольку не все продукты, изготовляемые в асептических условиях, могут иметь контейнер в качестве окончательной упаковки, в настоящем стандарте также используется выражение "форма продукта/контейнера".
2.14 критическая производственная зона (critical processing zone): Локальная зона в асептическом производстве, в которой продукт и поверхности, контактирующие с продуктом, открыты в окружающую среду.
Примечание - Асептические операции, выполняемые в критической производственной зоне, могут включать асептические соединения, наполнение, надевание пробок и укупорку.
2.15 критическая поверхность (critical surfaces): Поверхность в критической производственной зоне, находящаяся в непосредственной близости к асептическим операциям и представляющая риск для продукта.
2.16 перепад давления (differential air pressure): Разность давлений между помещениями или зонами или внутри них.
2.17 дезинфицирующее средство (disinfectant): Химический или физический агент, который инактивирует вегетативные микроорганизмы, но необязательно высокорезистивные споры.
2.18 микрофлора окружающей среды (environmental flora), изоляты окружающей среды: Микроорганизмы, присутствующие в технологической или производственной среде и/или выделенные в ней.
2.19 газовый фильтр (gas filter): Пористый материал, устанавливаемый в линию со сжатым воздухом с целью удаления неживых и/или живых частиц из газовых потоков, которые идут прямо или косвенно в зону контакта с продуктом.
2.20 медицинская продукция - продукт (health care product): Медицинские изделия, лекарственные средства (фармацевтические или биологические) и диагностические средства "in vitro".
2.21 высокоэффективный фильтр очистки воздуха по частицам (high efficiency particulate air filter), HEPA фильтр-ХЕПА фильтр: Устройство, в котором с помощью фильтрующего материала осуществляется отделение аэрозольных частиц от фильтруемого воздуха с минимальной эффективностью 99,97% (что соответствует коэффициенту проскока 0,03%) для частиц с размером 0,3 мкм DOP (диоктилфталат) - аэрозоля или другого предусмотренного документацией аэрозоля.
2.22 ламинарный поток воздуха (laminar air flow): Поток воздуха, в котором скорости воздуха вдоль параллельных линий тока одинаковы.
Примечание - Ламинарный поток части используется в ламинарных шкафах и укрытиях.
См. также однонаправленный поток (2.33).
2.23 наполнение средами - имитация наполнения (media fills): Метод оценки асептического процесса с использованием питательной среды для микроорганизмов.
Примечание - Наполнение средами - это синоним имитации процесса для контроля, имитатора наполнения продуктом, имитации процесса наполнения, питательного бульона для испытаний, наполняемого питательного бульона и т.д.
2.24 другие производственные зоны (other processing zones): Производственные зоны, не относящиеся к критическим производственным зонам, в которых медицинские продукты не открыты для окружающей среды.
Примечание - Эти зоны включают зоны подготовки, транспортирования и хранения стерилизованных компонентов, контейнеров и балк-продукта (нерасфасованного продукта - "ангро") в защитной таре; выгрузочные зоны автоклавов и производственные помещения, из которых производится доступ в критические зоны.
2.25 контактная поверхность продукта (product contact surfaces): Поверхность, которая приходит в контакт со стерилизованным продуктом или контейнером/укупоркой.
2.26 стерилизующий фильтр для продукта (product sterilizing filter): Пористый материал с номинальным размером пор 0,22 мкм, способный удерживать определенное количество микроорганизмов при определенных условиях и с использованием определенных методов контроля.
2.27 аттестация (qualification): Документированный исследовательский процесс, используемый изготовителем медицинской продукции, чтобы удостовериться в надежности и свойствах оборудования и/или процесса, проводимый до их приемки к использованию в производстве.
Примечание - Аттестация оборудования и/или процессов обычно включает в себя аттестацию установленного оборудования (после монтажа), аттестацию в оснащенном состоянии, аттестацию в эксплуатации.
2.27.1 аттестация установленного оборудования (installation qualification): Процесс, который показывает, что установка или процесс калиброваны и соответствуют всем относящимся к ним проектным критериям и стандартам безопасности.
2.27.2 аттестация в оснащенном состоянии (operational qualification): Комплекс проверок, который демонстрирует, что оборудование и/или процесс функционируют как предусмотрено, существуют инструкции по эксплуатации оборудования и персонал обучен включать, управлять и обслуживать оборудование.
2.27.3 аттестация в эксплуатации (performance qualification): Комплекс проверок системы, доказывающий ее эффективность и воспроизводимость.
2.28. смена (shift): Плановый период работы или производства, продолжительность которого обычно не превышает 12 ч, в котором занята одна и та же группа работников.
2.29 стерильный (sterile): Состояние, свободное от живых микроорганизмов.
Примечание - На практике не существует абсолютно надежного способа доказательства отсутствия живых микроорганизмов (см. 2.30).
2.30 стерилизация (sterilization): Валидированный процесс, обеспечивающий отсутствие живых микроорганизмов в продукте.
Примечание - Число микроорганизмов, которые выживают после процесса стерилизации, может быть выражено в вероятностном виде. Вероятность может быть снижена до очень малого значения, но никогда не может быть снижена до нуля.
2.31 вспомогательные зоны вне зоны асептического производства ЗАП (support areas outside АРА): Зоны с контролем окружающей среды, не находящиеся внутри асептической зоны производства и не являющиеся частью критической или иной производственной зоны.
2.32 финишная стерилизация (terminal sterilization): Процесс, при котором продукт стерилизуется в окончательной упаковке и который позволяет проводить измерения и количественную оценку летальности микроорганизмов.
2.33 однонаправленный поток воздуха (unidirectional air flow): Поток воздуха, линии тока которого имеют одно направление и который может обладать или не обладать одинаковыми скоростями струй воздуха по ходу течения параллельных линий тока.
См. также ламинарный поток воздуха (2.22).
2.34 вентиляционный фильтр (vent filter): Пористый материал, способный удерживать живые и неживые частицы из газов, входящих в закрытый объем и выходящих из него.
3 Система управления качеством
Для того, чтобы обеспечить контроль за всеми действиями, влияющими на асептическое производство, должна быть организована система управления качеством, соответствующая характеру выполняемых операций. Если не применяются заменяющие это понятие национальные, региональные или международные Правила надлежащего производства - Good Manufacturing Practice, система управления качеством должна соответствовать требованиям ГОСТ Р ИСО 9001* и/или ГОСТ Р ИСО 9002* [5, 6].
________________
* На территории Российской Федерации действует ГОСТ Р ИСО 9001-2001, здесь и далее. - Примечание.
Примечание - В дополнение к требованиям к продукции, компонентам и производственным регламентам система управления качеством может включать письменные инструкции и требования в отношении:
a) условий окружающей среды в зоне асептического производства;
b) очистки и дезинфекции зоны асептического производства;
c) стерилизации продукта, оборудования и системы контейнер/укупорка;
d) асептического производства балк-продукта (нерасфасованного продукта), например лиофилизации, асептической кристаллизации, сушки порошков и т.д.;
e) перемещения различных предметов в зону асептического производства или критическую производственную зону;
f) порядка переодевания персонала;
g) внутрипроизводственного контроля и оценки;
h) обучения операторов и технического персонала;
i) порядка контроля за изменениями;
j) валидации.
4 Персонал
4.1 Управление персоналом
4.1.1 Следует разработать и принять инструкции по эксплуатации асептического производства, включая обучение персонала и контроль.
4.1.2 Следует контролировать эффективность использования этих инструкций.
4.1.3 Управленческий персонал должен отвечать за соответствие обучения требованиям, необходимым для допуска персонала к работе в асептической зоне в соответствии с 4.2.
4.2 Обучение для аттестации на право работать в зоне асептического производства
4.2.1 Весь персонал, имеющий допуск в зону асептического производства, должен быть аттестован на соответствие требованиям 4.2.2 и 4.2.3. Обучение по различным дисциплинам и выполняемым работам следует проводить с учетом индивидуальных обязанностей и направлено на достижение требуемого уровня знаний.
4.2.2 Критерии оценки соответствия персонала должны быть отражены в учебных программах. Весь персонал, работающий в зонах асептического производства, в том числе занятый техническим обслуживанием, должен быть обучен следующему:
a) гигиене персонала, например, мытью рук и методам дезинфекции;
b) правилам, касающимся ювелирных изделий или применения косметики;
c) асептической технике, например, персонал, работающий в зоне асептического производства, должен избегать:
1) лишних движений и контактов с критическими поверхностями;
2) лишних движений и разговоров, которые могут вызвать генерацию частиц и турбулентность;
3) движений над открытыми контейнерами и открытым продуктом и компонентами;
4) создания препятствий движению воздуха над критическими поверхностями;
d) основам микробиологии;
e) процедурам переодевания (см. раздел 9);
f) производству стерильных продуктов внутри зоны асептического производства;
g) действиям по защите качества продукции в экстренных случаях, например, при отказах системы вентиляции и кондиционирования воздуха, системы энергоснабжения и пр.
4.2.3 Персонал, имеющий периодический доступ в зону асептического производства, включая руководителей, работников служб контроля и обеспечения качества, который имеет периодический доступ в зону асептического производства, должен быть обучен и аттестован в соответствии с 4.2.2 и должен быть обучен следующим основным вопросам:
a) гигиене персонала;
b) правилам, касающимся ювелирных изделий или применения косметики;
c) основным элементам асептической техники;
d) основам микробиологии;
f) процедурам переодевания.
Если с учетом строгих ограничений зоны асептического производства это трудно осуществить на практике, то указанный персонал должен тщательно контролироваться опытными работниками.
4.2.4 Следует вести документацию о прохождении обучения и аттестации.
4.2.5 Весь персонал, который непосредственно принимает участие в операциях наполнения или производства стерильной продукции в критических зонах, должен принимать участие в испытаниях с использованием наполнения средами в соответствии с требованиями настоящего стандарта не менее одного раза в год.
4.2.6 Новый персонал, работающий в критических производственных зонах, должен принять участие не менее чем в одном испытании с использованием наполнения средами или эквивалентной асептической операции, которая может производиться в учебной обстановке, до того как он будет допущен принять участие в работе, выполняемой в критических производственных зонах.
4.2.7 Весь персонал должен проходить повторное обучение в соответствии с документированными инструкциями как по выполняемым функциям, так и по соответствующим элементам системы обеспечения качества с определенной периодичностью или при необходимости.
4.3 Общее состояние здоровья персонала
4.3.1 Персонал должен сообщать об изменениях состояния своего здоровья, которые могут повлиять на асептическое производство (воспаление, повреждения кожи, обычная простуда, понос и пр.).
4.3.2 При изменении состояния здоровья, влияющем на работу в асептическом производстве, работники не должны допускаться в критические производственные зоны, но могут работать в других зонах.
Примечание - Персонал, допускаемый для работы в асептических условиях, должен проходить первичное и периодическое медицинские обследования.
4.4 Контроль персонала
4.4.1 Работники, обученные и аттестованные для работы в зоне асептического производства, должны проходить микробиологический контроль по соответствующей программе, которая включает отбор проб с одежды и перчаток.
Примечание - Общим правилом является проведение микробиологического отбора проб с одежды и перчаток после их использования.
4.4.2 Результаты этого контроля должны использоваться для обнаружения тенденций изменения контаминации и оценки необходимости повторного обучения персонала.
5 Проектирование помещений
5.1 Особенности проектирования помещений
При проектировании зон асептического производства нужно учесть следующие особенности планировочных решений и конструкций:
а) обеспечить приспособленность поверхностей стен, пола и потолка для очистки и их способность выдерживать обработку дезинфицирующими средствами;
b) эффективно герметизировать потолки;
c) избегать уступов и других горизонтальных поверхностей, на которых могут скапливаться частицы и которые могут нарушать потоки воздуха;
d) выполнять монтаж трубопроводов, воздуховодов и прочих коммуникаций так, чтобы избежать образований труднодоступных мест или других поверхностей, трудно доступных для очистки;
e) предусматривать достаточно места для зон переодевания, хранения чистой и загрязненной одежды и мытья рук;
f) разделять зоны переодевания и подготовки от зоны асептического производства посредством воздушных шлюзов и передаточных камер для компонентов, материалов и оборудования;
g) учитывать характер потоков воздуха, которые могут повлиять на продукт и критические поверхности;
h) устанавливать окна и другие средства наблюдения, где это нужно;
i) поддерживать соответствующий перепад давления воздуха между помещениями различных классов;
j) оборудовать воздушные шлюзы системами, исключающими нахождение обеих дверей в открытом состоянии;
k) поддерживать температуру и, если необходимо, относительную влажность в допустимых пределах и, по возможности, с непрерывным контролем;
l) располагать оборудование в зоне асептического производства таким образом, чтобы облегчить доступ к нему оператора и обслуживающего персонала и свести до минимума возможность сообщения открытых контейнеров и продукта с окружающей средой;
m) располагать оборудование, требующее частого вмешательства оператора или обслуживающего персонала, в удалении от критических производственных зон;
п) учитывать потенциальные источники перекрестного загрязнения.
Примечания
1 Особое внимание должно быть уделено выбору места расположения зоны асептического производства относительно других зон в производственном здании. Обоснование выбора этого места должно быть документировано.
2 В зданиях многоцелевого назначения зону асептического производства следует располагать вдали от зон с интенсивными потоками транспорта (материалов, оборудования и персонала) или отделять ее физическими барьерами.
3 Если в зоне асептического производства используются чувствительные вещества, цитотоксические или другие опасные материалы, то в проекте помещения эти особенности должны быть учтены.
5.2 Рассмотрение проекта помещений
5.2.1 Процесс рассмотрения проекта помещений должен быть проведен и документирован так, чтобы показать соответствие проекта специфическим требованиям производства продукции. Проект помещения следует рассматривать также при введении новых процессов или видов продукции.
5.2.2 Для существующих устройств следует провести ретроспективный анализ этого требования.
5.3 Поток материалов
В проекте помещений, где располагается зона асептического производства, следует предусмотреть контроль за потоками компонентов и материалов, чтобы:
a) поддерживать микробиологическую чистоту в критических производственных зонах;
b) минимизировать попадание загрязнений в зону асептического производства и обеспечить контроль загрязнений таким образом, чтобы не допустить их в критические производственные зоны;
c) исключить смешивание чистых и грязных предметов.
6 Зоны асептического производства
6.1 Общие положения
Примечание - Зона асептического производства состоит из зон, для которых требуется разделение и контроль. Требования к качеству воздуха в каждой зоне зависят от вида выполняемых операций. Эти зоны разделяются на критические производственные зоны и прочие производственные зоны.
6.1.1 Зона асептического производства должна представлять собой контролируемое пространство, в котором загрязнение микроорганизмами и частицами находится в установленных пределах.
6.1.2 Доступ в зону асептического производства должен быть ограничен; в нее может входить только аттестованный персонал, как указано в 4.2.
6.1.3 Следует предусмотреть необходимый однонаправленный поток воздуха и/или положительный перепад давления, чтобы не допустить попадания загрязнений в зону асептического производства из соседних зон.
6.1.4 Должна быть принята, документально оформлена, введена и поддерживаться программа контроля окружающей среды.
6.2 Критические производственные зоны
6.2.1 Должны быть определены критические производственные зоны и документально установлены требования к допустимому загрязнению этих зон микроорганизмами и частицами.
6.2.2 Должны быть предприняты необходимые меры по сведению к минимуму возможности загрязнения стерилизуемых предметов, материалов или окружающей среды.
6.2.3 В 1 м![]() воздуха критических производственных зон должно содержаться менее 3500 частиц размером 0,5 мкм и более в состоянии "в эксплуатации".
воздуха критических производственных зон должно содержаться менее 3500 частиц размером 0,5 мкм и более в состоянии "в эксплуатации".
Примечания
1 Качество воздуха должно соответствовать зоне А, классу 5 ИСО или классу 100 в используемых национальных и международных стандартах по качеству воздуха.
2 Критические производственные зоны подлежат эффективному контролю во время эксплуатации с целью выявления тенденций изменения параметров окружающей среды.
3 Допускается, что не всегда возможно проводить контроль соответствия чистоты по частицам во время процесса наполнения из-за образования частиц или капелек от самого процесса.
6.2.4 Критические производственные зоны подлежат текущему контролю на присутствие микроорганизмов, т.е. микрофлоры/изолятов в окружающей среде (14.3.1.1) и аэрозольных частиц (14.4).
6.3 Другие производственные зоны (внутри зоны асептического производства)
6.3.1 Должны быть определены другие производственные зоны и установлены требования к их загрязнению микроорганизмами и частицами.
6.3.2 Должны быть предприняты необходимые меры по сведению к минимуму возможности загрязнения стерилизуемых предметов, материалов или окружающей среды.
6.3.3 В 1 м![]() воздуха других производственных зон должно содержаться менее 350000 частиц размером 0,5 мкм и более в состоянии "в эксплуатации".
воздуха других производственных зон должно содержаться менее 350000 частиц размером 0,5 мкм и более в состоянии "в эксплуатации".
Примечание - Это качество воздуха обычно соответствует зоне В, классу 7 ИСО или классу 10000 в используемых национальных и международных стандартах по качеству воздуха.
6.3.4 Другие производственные зоны подлежат текущему контролю на присутствие микроорганизмов, то есть микрофлоры/изолятов в окружающей среде (14.3.1.1) и аэрозольных частиц (14.4).
7 Вспомогательные зоны за пределами зоны асептического производства
7.1 В 1 м![]() воздуха вспомогательных зон должно содержаться менее 3500000 частиц размером 0,5 мкм и более (состояние "оснащенное" или "в эксплуатации" специфицируется в документации)*.
воздуха вспомогательных зон должно содержаться менее 3500000 частиц размером 0,5 мкм и более (состояние "оснащенное" или "в эксплуатации" специфицируется в документации)*.
_______________
*В ИСО 13408-1 указано только состояние "в эксплуатации". В Правилах GMP ЕС рассматривается как состояние "в эксплуатации", так и "оснащенное" состояние. Это учтено в настоящем стандарте.
Примечания
1 Это качество воздуха должно соответствовать зонам С/D, классам 7 (8) ИСО или классам 10000 (100000) в используемых национальных и международных стандартах по качеству воздуха.
2 Дезинфекция и контроль окружающей среды этих зон могут быть менее частыми, чем для производственных зон.
7.2 Вспомогательные зоны подлежат текущему контролю на присутствие микроорганизмов, то есть микрофлоры/изолятов в окружающей среде.
8 Системы вентиляции и кондиционирования воздуха и контроля
Примечание - Основные элементы системы вентиляции и кондиционирования воздуха и программы контроля требуют соответствующего подхода при проектировании и контроле асептических производственных зон, включая обеспечение относительной влажности, температуры, скорости потока воздуха, высокоэффективной фильтрации, организации однонаправленных потоков воздуха и перепада давления между соседними помещениями.
8.1 Температура и относительная влажность
8.1.1 Значения температуры и, при необходимости, относительной влажности воздуха должны быть определены, контролироваться и регулироваться так, чтобы выполнялись требования комфорта сотрудников и качества продукта. Это прямо влияет на асептическую технологию и потенциальный уровень загрязнений.
8.1.2 Эти значения должны учитывать требования работающего персонала и эксплуатируемого оборудования.
8.2 Воздух
8.2.1 Перепад давления воздуха между соседними зонами должен быть специфицирован, контролироваться и регулироваться.
8.2.2 Во все критические производственные зоны должен подаваться воздух от НЕРА фильтров, скорость которого должна соответствовать требованиям установленного класса чистоты.
8.2.3 Скорость однонаправленного потока воздуха должна контролироваться для каждого НЕРА фильтра в соответствии с программой через определенные интервалы времени.
Примечание - Значительное снижение скорости потока воздуха может привести к увеличению загрязнения.
8.2.4 Должны быть определены и документированы требования к потокам воздуха, чтобы показать соответствие потока воздуха технологическому процессу; следует провести исследования влияния турбулентности на эффективность очистки потоком воздуха.
8.3 Целостность НЕРА фильтров
8.3.1 Сертификация
Поставщик должен представить сертификат на НЕРА фильтры, подтверждающий их эффективность.
8.3.2 Первоначальный и периодический контроль
8.3.2.1 Целостность фильтров должна быть проверена соответствующим методом после их установки.
8.3.2.2 Когда необходимые параметры поддерживаются за счет НЕРА фильтров, последние должны быть проверены после установки и аттестованы при помощи соответствующего теста, например, холодного или горячего DOP или теста, парафинового масла, химического аэрозоля и пр.
8.3.2.3 Скорость потока воздуха, проходящего через фильтры, должна проверяться с определенной периодичностью.
8.3.2.4 После любого изменения конфигурации потока воздуха должна быть заново проведена его проверка.
8.3.2.5 Фильтры должны проверяться согласно документированной методике после возникновения любой ситуации, которая может повлиять на целостность фильтров, или когда результаты контроля окружающей среды указывают на возможность ухудшения целостности фильтров.
9 Переодевание
9.1 Обучение порядку переодевания
9.1.1. Персонал должен быть обучен соответствующему порядку переодевания, чтобы риск внесения загрязнения в зону асептического производства был минимальным.
9.1.2 Эффективность обучения должна быть проверена с помощью микробиологических методов.
Примечание - С целью демонстрации эффективности переодевания могут использоваться также общие методы оценки загрязнения частицами.
9.1.3 Результаты проверки умения переодеваться должны быть доведены до сведения работников.
9.2 Требования к переодеванию
9.2.1 Персонал должен носить специальную одежду (первого переодевания), включая обувь, до того, как он войдет в зону переодевания.
Примечания
1 Персонал должен входить в зону переодевания через воздушный шлюз.
2 Персонал может переодеваться в специальную одежду предприятия (первое переодевание) в воздушном шлюзе, находящемся в близлежащем помещении по отношению к зоне переодевания.
9.2.2 Во вспомогательных зонах персонал должен носить одежду, которая отвечает требованиям чистоты по частицам, установленным для этих зон.
Примечание - Головной убор и, если требуется, маска для бороды надеваются в воздушном шлюзе. Одноразовые бахилы могут использоваться в дополнение к специальной обуви.
9.2.3 Персонал, входящий в зону асептического производства, должен носить одежду, обработанную так, чтобы на ней отсутствовали микроорганизмы и выделение частиц было минимальным.
Примечания
1 Стерильные пластиковые очки часто надеваются поверх капюшона для защиты глаз и бровей. Следует тщательно следить за тем, чтобы в районе лодыжек ног, запястий или шеи не было промежутков или открытых участков кожи. При выполнении в чистых помещениях некоторых операций используются нарукавники, высокие бахилы и двойные перчатки, чтобы свести до минимума вероятность появления незакрытых участков или неплотностей при движении.
2 Одежду, проверенную на наличие микробного загрязнения (см. 4.4), можно использовать в зоне асептического производства только после соответствующей обработки.
9.3 Контроль одетого персонала
9.3.1 Порядок контроля того, что персонал не наносит вреда окружающей среде в зоне асептического производства, должен быть определен письменными инструкциями.
9.3.2 Перчатки и одежду следует систематически проверять на соответствующее прилегание и целостность.
9.3.3 Работникам, занятым во вспомогательных зонах вне зоны асептического производства, запрещается доступ в зону асептического производства без соответствующего обучения и переодевания (см. 4.2).
Примечание - Персонал, связанный с операциями наполнения, не должен в течение смены заменяться работниками, выполняющими другие функции внутри зоны асептического производства.
10 Очистка и дезинфекция зоны асептического производства
10.1 Дезинфицирующие и моющие средства
10.1.1 Следует проводить систематическую очистку и дезинфекцию асептических производственных зон в соответствии с документированными инструкциями.
10.1.2 Следует использовать только валидированные и разрешенные к использованию моющие и дезинфицирующие средства.
10.1.3 Моющие и дезинфицирующие средства не должны содержать микробного загрязнения.
10.1.4 Следует оформлять и сохранять протоколы о проведении очистки и дезинфекции.
10.1.5 Инструкции должны включать данные об используемых разрешенных средствах, графиках очистки, применении дезинфицирующих средств, очистки после дезинфекции, если это требуется, мерах по защите персонала, требованиях к хранению моющих и дезинфицирующих средств и мерах предосторожности.
10.1.6 Следует провести валидацию методов удаления остатков моющих и дезинфицирующих средств с поверхностей, которые контактируют с продуктом.
10.1.7 Дезинфицирующие средства должны иметь установленный срок годности.
Примечания
1 Емкости, в которых хранятся дезинфицирующие средства, должны быть тщательно очищены и, если необходимо, стерилизованы перед повторным использованием.
2 При хранении и использовании применяемых средств нужно строго следовать инструкциям изготовителя.
3 При выборе дезинфицирующих средств и методов их применения следует принимать во внимание национальные требования безопасности персонала.
4 Следует предусматривать смену или ротацию дезинфицирующих средств, исходя из возможных изменений в микрофлоре/изолятах помещений.
5 Если контроль окружающей среды указывает на присутствие спорообразующих микроорганизмов, плесени и грибов, может потребоваться применение спороцидных агентов.
6 Емкости для дезинфицирующих и моющих средств и другое оборудование для очистки зон асептического производства должны использоваться только в этих зонах.
10.2 Валидация методов дезинфекции
10.2.1 Эффективность и периодичность проведения дезинфекции должны быть определены при валидации.
10.2.2 Для каждого помещения и оборудования должны быть разработаны и находиться на месте соответствующие инструкции по оценке, аттестации и контролю использования дезинфицирующих средств.
Примечание - Оценку эффективности дезинфицирующих средств следует проводить с учетом уменьшения типов и количества микроорганизмов, выявленных на поверхностях во время текущего контроля окружающей среды.
10.3 Контроль эффективности очистки и дезинфекции
10.3.1 Оценка эффективности очистки и дезинфекции должна быть составной частью общей программы контроля окружающей среды.
10.3.2 При появлении необычных результатов микробиологического контроля или необычной устойчивости микроорганизмов нужно провести и документально оформить исследование по обнаружению источника загрязнения.
11 Аттестация оборудования, используемых средств и валидация процесса
11.1 Общие положения
11.1.1 Все выполняемые в асептической производственной зоне процессы, которые влияют на стерильность или качество продукта, должны быть валидированы.
11.1.2 Должны быть разработаны инструкции, описывающие все операции в критической зоне (критическом оборудовании).
11.2 Аттестация технологического оборудования
Технологическое оборудование (стерилизаторы, средства очистки, фильтры, установки наполнения, оборудование по укупорке, оборудование для герметизации и установки лиофильной сушки и т.п.) должно быть аттестовано на соответствие своему назначению.
11.3 Валидация стерилизации поверхностей оборудования, контактирующих с продуктом
11.3.1 Поверхности оборудования, контактирующие с продуктом, должны быть стерилизованы.
11.3.2 Процессы стерилизации, относящиеся к поверхностям вышеуказанного оборудования, должны быть валидированы.
11.4 Валидация средств, используемых в технологическом процессе
Средства, используемые в технологическом процессе, такие как вода очищенная, вода для инъекций, сжатый воздух для фармацевтических целей (и/или другие газы), чистый или приготовленный на основе воды для инъекций пар, а также системы очистки на месте и обработки паром на месте должны быть валидированы на предмет соответствия своему назначению.
12 Материалы и оборудование, поступающие в зону асептического производства
12.1 Стерилизация
Каждый процесс стерилизации компонентов или материалов, используемых в асептической зоне, подлежит независимой валидации.
12.2 Газы
12.1.1 Сжатые газы, которые контактируют с продуктом, контейнером, упаковкой или поверхностью, соприкасающейся с продуктом, подлежат стерилизации.
12.1.2 Сжатый воздух следует контролировать на влажность и загрязнение маслами.
12.3 Биозагрязнения
Следует периодически определять характеристики и резистентность к микробной инактивации биозагрязнений на поверхностях компонентов и оборудования, которые поступают в зону асептического производства.
12.4 Депирогенизация
При использовании процесса депирогенизации должно быть показано, что этот процесс может удалять большее количество эндотоксинов, чем их может изначально присутствовать в компоненте или продукте.
Примечания
1 Следует установить количество эндотоксинов на материалах до проведения процесса депирогенизации.
2 Медицинские изделия и/или контейнеры из пластмассы могут проходить депирогенизацию полосканием (промыванием), и/или при высокотемпературном литье и/или экструзии до наполнения. Эти предметы должны быть стерилизованы как можно скорее после мойки, чтобы предотвратить загрязнение эндотоксинами.
3 Резиновые пробки могут считаться свободными от пирогенов за счет многократных циклов мытья и полоскания перед окончательной стерилизацией паром.
13 Производство
13.1 Следует контролировать смешивание нерасфасованных растворов, чтобы предупредить повышение уровня загрязнения микроорганизмами и, возможно, эндотоксинами, которые могут появиться к этому времени, до того как нерасфасованный раствор пройдет стерилизующую фильтрацию.
Примечание - Смешивание растворов должно проводиться во вспомогательных зонах (см. 7.1) в герметизируемых резервуарах, особенно если раствор должен храниться до фильтрации.
13.2 Общее время, затрачиваемое на фильтрацию продукта и наполнение им, а также время от окончания фильтрации до начала наполнения не должно превышать установленного максимально допустимого предела.
Примечание - Время между операциями мытья и стерилизации должно быть минимальным и не превышать установленного максимально допустимого предела.
13.3 Все технологические процессы перемещения и переработки материалов и движения оборудования, поступающих в асептические зоны, должны быть оформлены документально.
14 Программа контроля окружающей среды
Примечание - В программах контроля окружающей среды приводится порядок текущего контроля за загрязнением частицами или микроорганизмами в производственных и других зонах, а также план мер, которые необходимо принять при превышении уровней действия.
14.1 Инструкции
14.1.1 Должны быть разработаны, документированы и выполняться инструкции по контролю окружающей среды.
14.1.2 Эти инструкции должны содержать:
a) периодичность контроля;
b) вид контроля;
c) места контроля;
d) уровни предупреждения и действия;
e) предпринимаемые действия при нарушении установленных требований.
14.2 Отбор проб
14.2.1 Производственная зона и периодичность отбора проб
14.2.1.1 Участки критической производственной зоны, где происходит контакт с продуктом и материалами, должны контролироваться в течение операции наполнения и/или немедленно после ее завершения.
14.2.1.2 Отбор проб в критических производственных зонах следует проводить так, чтобы риск загрязнения продукта был минимальным.
14.2.1.3 Контроль других производственных зон следует проводить с определенной периодичностью, основанной на классификации зон и данных контроля.
14.2.1.4 Вспомогательные зоны вне ЗАП следует контролировать с определенной периодичностью, но контроль в них может производиться реже, чем в производственных зонах.
14.2.1.5 Периодичность такого контроля для различных помещений и оборудования должна основываться на данных контроля окружающей среды в прошедший период с учетом вида продукции и процесса.
14.2.2 Места отбора проб
Места отбора проб должны быть теми же, что и при проведении валидации.
Примечание - Конкретные места отбора проб для каждой программы выбирает производитель с учетом различий в проекте помещений, конструкции оборудования и параметров процесса.
14.3 Программа микробиологического контроля окружающей среды
14.3.1 Общие положения
14.3.1.1 Зона асептического производства подлежит регулярному микробиологическому контролю, т.е. контролю микрофлоры/изолятов окружающей среды. Периодический контроль должен включать в себя соответствующие методы для дрожжей, плесеней и других микроорганизмов.
14.3.1.2 Должны использоваться валидированные методы выявления микроорганизмов и калиброванное оборудование.
14.3.1.3 Следует отбирать пробы в зонах, в которых компоненты и продукт открыты для окружающей среды, например, в критических производственных зонах и линиях наполнения.
14.3.1.4 Следует проводить дополнительный контроль загрязненности по микроорганизмам и частицам после начала эксплуатации или после периодов длительного отключения или модернизации оборудования и помещения.
14.3.1.5 Газы, которые проходят стерилизующую фильтрацию и контактируют с продуктом, первичной упаковкой или поверхностями, имеющими прямой контакт с продуктом, должны периодически контролироваться на присутствие микроорганизмов.
14.3.2 Дополнительные микробиологические характеристики
Программа микробиологического контроля окружающей среды должна включать периодическое определение характеристик микрофлоры/изолятов окружающей среды, чтобы оценить риск для продукта.
14.3.3 Методы отбора микробиологических проб
Отбор определенного объема проб воздуха и другие методы отбора проб, например, методы седиментационных пластин, смывов и контактных пластин, должны использоваться соответствующим образом, чтобы оценить микробную загрязненность внутри зоны асептического производства.
14.4 Программа контроля загрязненности окружающей среды частицами
Программы контроля загрязненности частицами должны разрабатываться для зон или оборудования, где частицы или условия окружающей среды могут влиять на качество продукта, безопасность персонала или достоверность контроля.
15 Уровни предупреждения и действия
15.1 Определение уровней предупреждения и действия
15.1.1 Уровни предупреждения и действия должны устанавливаться для всех точек пробоотбора в зоне асептического производства.
Примечания
1 Уровни предупреждения и действия должны устанавливаться исходя из результатов, полученных при валидации асептического процесса. Предшествующие данные текущего контроля также могут использоваться при установлении уровней предупреждения и действия.
2 Во многих случаях используется один уровень, который выполняет роль как уровня предупреждения, так и уровня действия для критических производственных зон.
3 Возможна корректировка уровней предупреждения и действия на основе периодических повторных оценок с использованием наполнения средами и соответствующих данных контроля окружающей среды.
15.2 Анализ данных
Результаты контроля окружающей среды должны своевременно анализироваться с учетом установленных для помещения и оборудования уровней предупреждения и действия. При этом следует проводить оценку влияния на качество продукта.
Следует предпринять соответствующие корректирующие действия, если результат контроля окружающей среды указывает на превышение установленных пределов.
15.3 Анализ тенденций изменения параметров окружающей среды
15.3.1 Следует постоянно проводить анализ тенденций изменения параметров окружающей среды.
15.3.2 Исходя из тенденций изменения параметров может потребоваться проведение исследования причин этого изменения.
16 Исследования и отчеты
16.1 Планы проведения исследований
16.1.1 Исследования необходимо проводить в случае нетипичных ситуаций, после крупных нарушений в работе или в случае превышения уровней действия.
16.1.2 Инструкции по проведению исследований должны включать определение или анализ:
a) данных, которые нужно собрать;
b) масштаба задачи;
c) влияния на продукт или контроля окружающей среды;
d) карантина для продукта;
e) необходимости проведения контроля окружающей среды;
f) последующей проверки;
g) уведомления привлекаемого ответственного персонала.
16.2 Исследовательский контроль
Должна быть разработана схема проведения исследовательского контроля, чтобы установить источник проблемы и показать, что зона или оборудование снова находятся под контролем. Может потребоваться проведение ревалидации исходя из результатов контроля.
16.3 Отчет об исследовании
16.3.1 Результаты исследования должны быть оформлены в виде отчета.
16.3.2 Отчет должен быть рассмотрен и утвержден руководством и распространен среди всего основного персонала, занятого в зоне асептического производства.
16.3.3 При необходимости отчет должен также содержать указания по корректирующим действиям в будущем и рекомендации по размещению продукта.
17 Наполнение средами (имитация процесса наполнения)
17.1 Контроль процесса
Для контроля за асептическими операциями необходимы хорошо обученный персонал, четкие инструкции, соответствующее оборудование и помещения. Наполнение средами в сочетании с надлежащим контролем окружающей среды может быть исключительно ценным для доказательства того, что асептическое приготовление стерильных растворов, суспензий и порошков выполняется требуемым образом. Наполнение средами должно имитировать асептический процесс настолько, насколько это целесообразно из практических соображений.
Примечание - Наполнение средами является этапом в процессе проверки характеристик системы асептического производства, включая окружающую среду, оборудование и персонал. Оно не дает автоматической гарантии того, что продукция, изготовленная на той же линии в другие периоды времени, будет иметь тот же уровень микробиологического качества. Однако путем контроля и валидации таких сопутствующих процессов, как мониторинг окружающей среды, аттестация персонала, очистка и стерилизация, можно поддерживать уровень качества продукции, продемонстрированный с помощью наполнения средами.
17.2 Первоначальная аттестация в эксплуатации
17.2.1 Аттестация в эксплуатации должна проводиться для каждой новой линии асептического наполнения и каждой новой формы (конфигурации) продукта/контейнера, которые не были представлены при предыдущей аттестации в эксплуатации.
17.2.2 Аттестация в эксплуатации должна включать исследование наполнения средами для формы продукта, которая является представительной для продукта/контейнера (упаковки), подлежащих наполнению. Представительные критерии включают в себя:
а) фактическую форму продукта/контейнера или форму, которая является представительной для других продуктов, наполнение которыми производится на данной линии наполнения;
b) два продукта, которые охватывают все остальные с учетом размера, наполнения, открывания упаковки, скорости линии, манипуляций и пр.;
с) продукт, который рассматривается как "наихудший случай" в плане возможности загрязнения, например, контейнера, который имеет наибольшее отверстие и перемещается вдоль линии с наименьшей скоростью.
17.2.3 Критерии приемлемости при аттестации в эксплуатации приведены в таблице 1.
Таблица 1 - Критерии приемлемости при наполнении средами для первоначальной аттестации в эксплуатации
| Размер серии в произ- | Минимальное число серий при испытаниях с наполнением средами | Мини- | УРОВЕНЬ ПРЕДУПРЕЖДЕНИЯ | УРОВЕНЬ ДЕЙСТВИЯ |
|
|
исходя из необходимости получить общее число наполненных единиц | 5000*' ** | Одна контаминированная единица в любой серии. Исследуется причина. Выполняется одна дополнительная серия. При неудаче в дополнительной серии аттестация в эксплуатации повторяется | Две контаминированные единицы в одной серии или по одной в каждой из двух серий. Аттестация с наполнением средами прекращается. Исследуется причина и повторяется первоначальная аттестация с наполнением средами |
| 500-2999 |
исходя из необходимости получить общее число наполненных единиц | 5000* | То же | То же |
|
| 3 (в каждой серии не менее 3000 единиц) | 9000 | Одна контаминированная единица в любой серии. Исследуется причина. Выполняется одна дополнительная серия. При неудаче в дополнительной серии аттестация в эксплуатации повторяется | Две контаминированные единицы в одной серии или по одной в каждой из двух серий. Аттестация с наполнением средами прекращается. Исследуется причина и повторяется первоначальная аттестация с наполнением средами |
| * Поскольку в каждой серии для испытаний с наполнением средами должно быть наполнено менее 3000 единиц, нельзя прямо применить данные таблицы 3. Однако, основываясь на накопленном опыте, можно считать отсутствие положительных единиц в каждой серии с наполнением средами как свидетельство низкого уровня загрязнения. | ||||
Число серий с наполнением средами и общее число наполненных единиц должно соответствовать следующим требованиям:
a) для серий продукции размером менее 500 единиц следует выполнить испытания не менее чем на трех сериях с наполнением средами и общим числом единиц, равным 5000;
b) для малых серий, редко выпускаемых продуктов или клинических серий (менее 500 упаковок, наполнение менее четырех раз в год), которые производятся на хорошо отлаженном аттестованном технологическом оборудовании в контролируемой окружающей среде, достаточно выполнить испытания на трех сериях с наполнением средами до наполнения продуктом или наполнением в клинических целях;
c) для серий размером от 500 до 2999 единиц испытания проводят не менее чем на трех сериях с наполнением средами и общим числом наполненных единиц, равным 5000;
d) для серий размером 3000 единиц и более испытания проводят не менее чем на трех сериях с наполнением средами с числом единиц 3000 в каждой серии и общим числом наполненных единиц, равным 9000.
Серии с наполнением средами по перечислениям а), b) и с) должны содержать число единиц, не меньшее, чем размер серии.
Примечание - Чтобы учесть колебания в процессе и вмешательства, происходящие в ходе текущего производства, может оказаться необходимым наполнять более 3000 единиц в серии с наполнением средами.
17.3 Периодические повторные аттестации
17.3.1 Плановые повторные аттестации с помощью наполнения средами должны проводиться не реже одного раза в 6 мес для каждой формы контейнеров (упаковки) и линии асептического наполнения.
17.3.2 Линии асептического наполнения и формы (виды) упаковки, используемые реже одного раза в 6 мес, должны проходить повторную аттестацию с помощью надлежащих тестов с наполнением средами.
17.3.3 Производство продукта может быть возобновлено до окончания процесса инкубации наполненных сред, но продукт не может быть реализован до того, как будут получены приемлемые результаты теста с наполнением средами.
Примечание - В случаях модификации оборудования, изменений в персонале, обнаружении аномалий при проверке параметров окружающей среды или при проверке на стерильность готового продукта может потребоваться повторная аттестация процесса или линии с помощью наполнения средами до истечения 6 мес.
17.3.4 Критерии приемлемости при повторной аттестации должны соответствовать требованиям таблицы 2. Число прогонов и общее число наполненных единиц должно соответствовать следующему:
a) для малых серий (менее 500 единиц) следует выполнять три прогона при максимальном размере серии;
b) в других случаях для малых серий, редко выпускаемых продуктов или клинических серий (менее 500 упаковок, наполнение менее четырех раз в год), должна допускаться повторная аттестация процесса или линии при выполнении одного прогона с наполнением средами с числом единиц, равным, по крайней мере, размеру серии, немедленно после наполнения серии продукции.
Примечание - Следует принять необходимые меры, обеспечивающие инактивацию и удаление остатков антимикробных веществ в линии наполнения до начала процесса наполнения;
c) при производстве серий от 500 до 2999 единиц следует выполнять один прогон наполнения средами с числом наполненных единиц не менее максимального размера серии;
d) при производстве серии размером 3000 единиц и более следует выполнять один прогон наполнения средами с числом наполненных единиц не менее 3000.
Примечание - Может оказаться необходимым наполнить более 3000 единиц, чтобы учесть колебания в процессе и вмешательства, происходящие в ходе текущего производства.
Таблица 2 - Критерии приемлемости при наполнении средами для периодической повторной аттестации
| Размер серии в произ- | Минимальное число серий при испытаниях с наполнением средами | Минимальное общее число наполненных единиц | УРОВЕНЬ ПРЕДУПРЕЖДЕНИЯ | УРОВЕНЬ ДЕЙСТВИЯ |
|
|
| Максимальный размер серии в производстве* | Одна контаминированная единица в любой серии. Повторная аттестация прекращается. Исследуется причина и выполняется периодическая аттестация | Две контаминированные единицы в одной серии. Повторная аттестация прекращается. Исследуется причина и повторяется первоначальная аттестация согласно таблице 1 |
| 500-2999 | 1 | Максимальный размер серии в производстве | же. Повторяется серия с наполнением средами | То же |
|
| 1 | 3000 | Повторяется серия с наполнением средами, если превышены значения уровней предупреждения по таблице 3 | См. таблицу 3 максимальных значений уровней действия |
| * Поскольку в каждой отдельной серии для испытаний наполнением средами должно быть наполнено менее 3000 единиц, нельзя прямо применить данные таблицы 3. Однако, основываясь на накопленном опыте, можно считать отсутствие положительных единиц в каждой серии с наполнением средами как свидетельство низкого уровня загрязнения. | ||||
17.4 Повторение первоначальной аттестации в эксплуатации
17.4.1 При необходимости следует повторить аттестацию в эксплуатации с использованием процедур, методов и критериев приемки по 17.1.
17.4.2 Следует повторить аттестацию в эксплуатации асептического процесса или линии наполнения, если:
a) превышен уровень действия (если только не установлено, что он из-за вызванной внешними обстоятельствами причины);
b) производственная линия не эксплуатировалась в течение длительного периода времени, например, одного года;
c) произошли существенные изменения.
Примечание - Инструкции по контролю изменений должны определять существенные изменения, такие как:
- изменения в оборудовании, непосредственно контактирующем с нерасфасованным продуктом или продуктом/контейнером;
- изменения в оборудовании или помещениях, которые могут оказать влияние на качество воздуха или воздушные потоки;
- существенные изменения в производственном персонале, например, создание новых бригад;
- образование дополнительных производственных смен.
17.5 Инструкции по проведению наполнения средами
17.5.1 Наполнение средами должно проводиться в отдельные дни и в различное время в течение обычного рабочего времени.
17.5.2 Следует иметь перечень допустимых и предписанных вмешательств во время проведения асептического процесса.
17.5.3 Наполнение средами должно проводиться в производственных условиях, включая условия "наихудшего случая", например, при наибольшем практически возможном числе допустимых вмешательств, устранении остановок линии, ремонте или замене игл или трубок, замене установленных в линии фильтров, изменении численности привлекаемого персонала и пр. Может потребоваться регулирование продолжительности цикла наполнения средами для специальных процессов, где происходят плановые прерывания процесса наполнения.
17.5.4 Наполнение средами должно иметь достаточную длительность, чтобы учесть влияние большинства манипуляций и действий, которые выполняются при нормальном ходе процесса в производстве.
Примечание - Если асептическому наполнению подлежит множество упаковок одной формы, но различных размеров, то можно отобрать определенные размеры для наполнения средами. Контейнеры с наиболее широким отверстием и наименьшая скорость движения линии должны быть включены в режим наполнения средами и могут представлять условия наихудшего случая. Однако в некоторых случаях маленькие упаковки представляют условия наихудшего случая из-за недостаточной стабильности контейнеров при работе линии и необходимости более частого ручного вмешательства.
17.6 Выбор сред и создание условий для роста микроорганизмов
17.6.1 После наполнения средами должен проводиться контроль роста микроорганизмов в средах, используемых для имитации процесса наполнения средами.
17.6.2 Температура инкубации должна быть такой же, как температура, используемая для единиц, наполненных средами.
17.6.3 Питательные среды, выбранные для наполнения средами, должны обеспечивать рост широкого спектра микроорганизмов, а также обеспечивать выявление и рост малого числа микроорганизмов, т.е. 100 КОЕ на единицу продукции или менее
Примечание - Результаты испытаний на рост тест-микроорганизмов должны соответствовать требованиям фармакопей. Такие испытания должны проводиться внутри контейнеров, наполненных средами, где это возможно.
17.7 Инкубация и контроль единиц, наполненных средами
17.7.1 Давшие течь или поврежденные единицы с имитирующими средами должны быть удалены. Об этом, согласно регламенту, должна быть сделана запись до начала инкубирования единиц.
17.7.2 Наполненные средами единицы должны инкубироваться не менее 14 сут.
17.7.3 Температуры инкубации должны соответствовать специфическим ростовым требованиям к росту тех микроорганизмов, присутствие которых предполагается в асептической зоне наполнения.
Примечание - Данные контроля окружающей среды могут использоваться при определении оптимальных температур инкубации. Наиболее часто используемые диапазоны температур инкубации составляют 20-25 °С, 30-35 °С или 28-32 °С.
17.7.4 Наполненные средой единицы должны сохраняться или обрабатываться так, чтобы обеспечивался контакт среды со всеми поверхностями, с которыми контактирует продукт.
17.7.5 После завершения инкубационного периода наполненные средой контейнеры должны быть визуально проверены на наличие роста микроорганизмов.
17.7.6 Микроорганизмы, находящиеся в контаминированных единицах, должны быть изучены с целью определения вероятного источника контаминации.
Примечания
1 Для предварительной оценки результатов может оказаться полезным контроль единиц в ранний период (3-7 сут инкубирования).
2 Следует регистрировать время наполнения единиц и время их анализа с целью облегчения определения времени, когда было проведено наполнение контаминированной единицы.
17.8 Критерии приемлемости
17.8.1 Общие положения
Риск загрязнения не должен превышать 0,1% при 95%-ном доверительном интервале для серии не менее 3000 единиц.
Примечания
1 Настоящий стандарт включает статистический подход к оценке вероятного риска контаминации при наполнении средами. Вероятность риска контаминации определяется по числу наполненных единиц и верхнему 95%-ному доверительному пределу (верхней границе 95%-ного доверительного интервала) нестерильных (положительных) единиц, вычисленному по числу фактически зафиксированных нестерильных единиц.
2 Хотя настоящий стандарт определяет риск контаминации производимой серии на уровне 0,1% при наполнении средами, производители должны стремиться к достижению меньшего риска. Автоматизированные линии быстрого наполнения и линии наполнения внутри изоляторов позволяют свести риск контаминации производимой серии продукции к значению менее 0,1%. Однако это трудно показать на практике, так как, например, для демонстрации степени контаминации 0,01% необходимо было бы наполнение средами 30000 единиц.
3 См. 17.2.3 и 17.3.4 для серий объемом менее 500 образцов.
Несмотря на значения, приведенные в таблице 3, все контаминированные единицы должны быть изучены с точки зрения причины и источника микроорганизмов, давших рост. Если возникает предположение, что производственная окружающая среда или продукт подвергаются риску контаминации, то независимо от числа обнаруженных микроорганизмов наполнение средами может быть расценено как неудачное.
17.8.2 Таблицы критериев приемлемости
17.8.2.1 В таблице 1 приведены критерии приемлемости для первоначальной аттестации в эксплуатации асептической линии наполнения.
17.8.2.2 В таблице 2 приведены критерии приемлемости для повторной аттестации асептической линии наполнения.
17.8.2.3 В таблице 3 приведены уровни предупреждения и действия, когда риск контаминации 0,1% получен для больших количеств единиц с наполнением средами, т.е. когда выбирается наполнение средами более 3000 единиц.
Уровни действия в таблице 3 основаны на риске контаминации 0,1%. Это значение получено делением числа наполненных средами единиц на верхний 95%-ный доверительный предел для наличия положительных (нестерильных) единиц согласно распределению Пуассона.
Таблица 3 - Уровни предупреждения и уровни действия для больших количеств единиц, наполненных средами
| Число единиц в одном испытании с наполнением средами | Уровень предупреждения* (число контаминированных единиц в одной серии с наполнением средами) | Уровень действия** (число контаминированных единиц в одной серии с наполнением средами) |
| 3000 | Не применяется | 1 |
| 4750 | 1 | 2 |
| 6300 | 1 | 3 |
| 7760 | 1 | 4 |
| 9160 | 1 | 5 |
| 10520 | 2 | 6 |
| 11850 | 2 | 7 |
| 13150 | 3 | 8 |
| 14440 | 3 | 9 |
| 15710 | 4 | 10 |
| 16970 | 4 | 11 |
| * Этот уровень предупреждения основан на выборе риска контаминации 0,05%. | ||
| Примечания | ||
Приложение А дает основополагающую информацию в отношении установления 95%-ного верхнего доверительного предела и установления 0,1%-ного уровня действия. В приложении А даны примеры вычисления частоты контаминации для данного количества единиц, наполненных средами.
Примечания
1 В таблице А.2 показано число контаминированных единиц в зависимости от числа единиц, наполненных средами. Таблица А.2 дает вспомогательные данные к таблице 3.
2 Таблица 3 и А.2 не предназначены для того, чтобы отдельные фирмы производили наполнение средами дополнительного числа единиц с целью получения большего числа приемлемых положительных единиц. Эти таблицы предназначены для демонстрации того, когда, по крайней мере, 0,1%-ный риск контаминации не достигнут.
Независимо от числа положительных (нестерильных) результатов, полученных при наполнении средами, производитель обязан исследовать источник микробной контаминации, чтобы обеспечить отсутствие риска как для окружающей среды асептического производства, так и для продукта.
17.9 Контаминация имитирующей средой
Контаминация помещения и оборудования имитирующей средой во время испытаний с наполнением средами не является основанием для вывода о непригодности для последующей работы этого помещения и оборудования. Имитирующая среда должна быть быстро удалена с последующей очисткой, дезинфекцией и, при необходимости, стерилизацией оборудования.
Примечание - При валидации методов очистки, дезинфекции и стерилизации должно быть продемонстрировано удаление пролитых сред, что может произойти при прогоне с наполнением средами.
17.10 Данные, необходимые для наполнения средами
Все испытания с наполнением средами должны быть полностью документированы, при этом для каждого испытания при необходимости должна быть дана следующая информация (или сделана ссылка):
a) дата и время наполнения средами;
b) наименование помещения, в котором проводятся испытания;
c) тип и размер контейнера/способа укупорки;
d) объем заполнения контейнера;
e) скорость наполнения;
f) серия и номер фильтра по каталогу;
g) вид наполняемой среды;
h) число наполненных единиц;
i) число отбракованных единиц при контроле и причина брака;
j) число инкубируемых единиц;
k) число нестерильных единиц;
l) время и температура инкубации каждой группы инкубируемых единиц и подвергается ли группа единиц двум различным температурным режимам инкубации;
m) методики, используемые для имитации любых стадий нормального процесса наполнения, которые могут включать, например, имитацию лиофилизации или замену газа для наполнения верхней части флакона;
n) данные микробиологического контроля при начале и во время испытаний с помощью наполнения средами;
о) список сотрудников, принимающих участие в испытаниях;
р) результаты роста микроорганизмов в имитирующих средах, извлеченных из наполненных контейнеров;
q) определение микроорганизмов из всех нестерильных единиц и исследование всех зафиксированных во время испытаний случаев контаминации;
r) отчет руководителя испытаний;
s) длительность периода времени хранения среды промежуточной емкости до фильтрации;
t) длительность времени, потребовавшегося для наполнения всех контейнеров (единиц).
17.11 Результаты испытаний с наполнением средами, превышающие уровни действия
17.11.1 Исследование
17.11.1.1 Когда уровень действия при наполнении средами превышен, должно быть проведено исследование и документирование причин такого превышения.
17.11.1.2 Если превышены уровни действия, то следует немедленно просмотреть все соответствующие записи, относящиеся к асептическому производству, между последним успешным наполнением и текущим наполнением.
Это исследование должно включать, по крайней мере, следующее:
a) данные микробиологического контроля окружающей среды;
b) данные контроля частиц;
c) данные контроля персонала (смывы с рук и т.д.);
d) режимы стерилизации имитатора, продукта и оборудования;
e) оценку НЕРА фильтров (уровень аэрозольного загрязнения частицами, тестирование с помощью дыма, измерение скоростей и т.д.);
f) направление воздушных потоков в помещении и перепад давлений;
g) подготовку и квалификацию оператора;
h) необычные явления, имевшие место во время испытаний;
i) условия хранения стерильных материалов и продукции;
j) определение контаминантов как ключ к источнику контаминации;
k) хозяйственные вопросы и обучение;
l) калибровка стерилизационного оборудования;
m) экспериментальные данные о чистоте воздуха перед фильтром и после него и/или о целостности корпуса фильтра;
n) недостатки продукта и/или технологического процесса; и/или ограничение процессов контроля;
о) документированная отбраковка проб по очевидным причинам до окончательных выводов.
17.11.2 Корректирующие действия
17.11.2.1 Если результаты испытаний с наполнением средами превышают уровни действия, то следует выполнить указания таблиц 1 и 2.
17.11.2.2 Решение о том, предпринимать ли действия в отношении произведенной и/или отправленной в реализацию продукции, должно исходить из оценки всей имеющейся информации и должно быть документировано.
17.11.2.3 Любой продукт, который произведен в линии на основе результатов испытаний с наполнением средами, должен быть изолирован до тех пор, пока не будут получены успешные результаты таких испытаний.
Примечание - Анализ производственных серий с учетом неудачных испытаний с наполнением средами включает соответствующие данные контроля окружающей среды, запись результатов испытаний на стерильность за этот период времени, возможные причины, объясняющие результаты текущих испытаний с наполнением средами, и любую другую информацию, которая может касаться стерильности рассматриваемого продукта.
18 Контроль стерильности готового продукта
18.1 Общие положения
Контроль стерильности асептически наполняемых продуктов должен проводиться для каждой серии (партии) продукта.
18.2 Исследование положительных (нестерильных) проб
18.2.1 Следует оценить положительные пробы по результатам контроля стерильности, а также провести исследования по установлению источника контаминации, включая рост микроорганизмов, вызванный контаминацией во время проведения контроля.
18.2.2 Следует оценить корреляцию между видами микроорганизмов, обнаруженными в производственной среде, помещениях лаборатории контроля стерильности, и видами микроорганизмов, выделенными при контроле стерильности.
18.3 Планы отбора проб
18.3.1 Планы отбора проб для оценки промежуточного или готового продукта должны быть разработаны с таким расчетом, чтобы обеспечить представительность каждой серии, и должны базироваться на статистическом обосновании или других приемлемых руководящих или регламентирующих требованиях.
18.3.2 План отбора проб должен быть представительным по отношению к серии.
Полученные пробы считаются представительными для всей серии, если они:
a) берутся периодически (включая начало, середину и конец процесса наполнения);
b) включают единицы, взятые после вмешательства оператора в производственную зону (зоны).
19 Обработка паром на месте
19.1 Общие положения
19.1.1 Оборудование, которое нельзя стерилизовать в автоклаве из-за больших размеров или особенностей конфигурации, стерилизуется на месте (in situ). При этом необходимо убедиться в том, что летальность процесса соответствует снижению на шесть десятичных логарифмов при использовании для первоначальной аттестации и повторной аттестации системы биоиндикаторов, устойчивых к воздействию влажного тепла.
Использование при валидации устойчивых к теплу микроорганизмов требует соответствующего контроля, чтобы минимизировать возможную контаминацию зоны асептического производства.
Обработка паром на месте позволяет выполнять стерилизацию паром всей технологической системы производства медицинской продукции как единого целого, исключив или уменьшив необходимость сборки в асептических условиях. Примерами могут служить резервуары, танки, линии наполнения, передаточные линии, системы фильтрации или системы подготовки воды для инъекций.
Процесс обработки паром на месте требует точного соблюдения процедур по удалению воздуха и конденсата, а также сохранения целостности системы после завершения обработки. Незначительные отклонения от установленной процедуры могут привести к нарушениям в технологическом процессе, которые могут остаться незамеченными. Нужно принять во внимание следующие меры при обработке паром на месте:
а) удаление воздуха гравитационным или вакуумным методами;
b) постоянную продувку (опорожнение) или паровые трапы (ловушки) во всех нижних точках с целью исключения образования конденсата;
c) строгое соблюдение процедур обработки паром на месте;
d) соответствующее поддержание целостности системы после обработки;
e) строгое соблюдение максимальных требований к фильтрам по температуре, давлению и потоку;
f) исключение случаев обратного давления на фильтрах во время обработки паром на месте.
19.1.2 Если отсутствует автоматическая система обработки паром на месте (SIP), то должны выполняться и документироваться ручные процедуры.
19.2 Целостность системы после стерилизации
19.2.1 После стерилизации должна поддерживаться целостность системы.
19.2.2 Газы (воздух или азот), вводимые в систему, должны быть стерильными.
19.2.3 Из системы должен быть удален пар и конденсат, и в системе должен поддерживаться положительный перепад давления до начала ее использования.
Примечание - Введение стерильного газа (например, воздуха или азота) может высушить систему до использования, что очень важно при производстве продуктов, не содержащих воду.
20 Фильтрация в технологическом процессе
20.1 Программа оценки фильтров и фильтровального оборудования
20.1.1 Документированная программа оценки фильтров должна быть разработана до проведения валидации и приемки фильтров.
Эта программа должна включать оценку следующего:
a) данных изготовителя о фильтрах и их элементах (мембранах, картриджах, вспомогательных элементах, колпачках, прокладках, кольцах и пр.);
b) инструкций по проверке целостности фильтров;
c) совместимости фильтра и продукта;
d) выделений из фильтра, в том числе выделений частиц.
20.1.2 Фильтры, предназначенные для удержания бактерий (стерилизующие фильтры), должны проверяться соответствующим и определенным биологическим тестом или должны иметь необходимые данные от изготовителя.
Примечание - Микробиологический контроль должен проводиться во время первоначальной аттестации в лабораторных условиях, если его проведение в производственных условиях может оказать отрицательное влияние на качество окружающей среды.
20.1.3 Следует оценить абсорбцию или адсорбцию фильтром лекарственного средства или консервирующего вещества.
20.2 Оценка безопасности фильтра и фильтрующей системы
20.2.1 Общие положения
Безопасность фильтра и фильтрующей системы следует оценивать биологическими и химическими методами, чтобы убедиться, что они не выделяют нежелательных материалов в проходящие через них газы или жидкости.
20.2.2 Биологические тесты на безопасность
20.2.2.1 Если в фильтре (фильтрующей системе) используются любые пластмассы, то для каждого вида пластмассы следует провести контроль на биологическую безопасность. Контроль может быть проведен для всего фильтра (фильтрующей системы). Следует произвести оценку фильтров на присутствие эндотоксинов.
20.2.3 Химические тесты
Для всех типов фильтров следует контролировать:
a) окисляющие субстанции;
b) влияние на стабильность в процессе хранения;
c) другие тесты, если требуется фильтрация раствора или газа со специальными свойствами.
20.3 Аттестация физической целостности в эксплуатации
20.3.1 Валидационная проверка физической целостности технологического фильтра должна производиться после его использования без нарушения корпуса и сборочных элементов фильтра.
В качестве основы для валидационного метода могут использоваться инструкции или рекомендации изготовителя.
Приемлемыми тестами на физическую целостность технологического фильтра являются "диффузный поток", "поддержание постоянного давления", "точка пузырька".
20.3.2 Следует определить свойство фильтра или его корпуса сохранять целостность при стерилизации и прохождении через него газа или жидкости (учитывая изменения давления или потока).
20.4 Фильтрация в линиях наполнения
20.4.1 Части фильтра и прокладки должны проверяться перед сборкой.
20.4.2 Сборка фильтр-пресса, дисковой мембраны или картриджа и корпуса должна проводиться в соответствии с инструкцией изготовителя фильтра.
20.4.3 В протоколе партии должен быть указан номер серии фильтра (от изготовителя).
21 Лиофилизация
21.1 Общие положения
Лиофилизация представляет собой более высокий этап в развитии асептической технологии. Несмотря на то, что лиофилизация представляет собой процесс, требующий комплексной технологии, настоящий стандарт рассматривает только аспекты, связанные с асептическим производством.
Лиофилизация должна проводиться в асептических условиях.
21.2 Валидация
21.2.1 Процесс лиофилизации должен быть валидирован и аттестован как составная часть общей валидации асептического процесса.
Технология лиофилизации может внести факторы, которые влияют на стерильность, активность, эффективность и стабильность готового продукта. Важную роль играют:
a) состав раствора;
b) наполнение во флаконы и валидацию операций наполнения;
c) стерилизация и технические вопросы, связанные с лиофильной установкой;
d) контроль готового продукта.
21.2.2 Валидации подлежат следующие элементы процесса лиофилизации:
a) передача продукта в лиофильную установку и из нее;
b) стерилизация лиофильной установки;
c) целостность лиофильной установки (тесты на утечку);
d) компьютерная система контроля (если применяется);
e) стерилизация передаточных кассет или противней;
f) дезинфекция передаточных устройств.
Примечание - Программа контроля, включая регулярные проверки с использованием наполнения средами или имитирующего питательного бульона, должна охватывать параметры валидации.
21.2.3 В случае существенных изменений в процессе должна быть снова проведена валидация (см. 17.2).
21.3 Параметры контроля процесса лиофилизации
Поскольку воздух из камеры удаляется, а затем вновь в нее подается во время процесса лиофилизации, места ввода патрубков удаления и подачи воздуха должны быть оснащены стерилизующими фильтрами. Если предусмотрена подача других газов (например, азота, углекислого газа) для сброса вакуума или защиты продукта, то пути прохождения этих газов также должны быть оснащены финишными стерилизующими фильтрами.
Чтобы показать способность оборудования контролировать и поддерживать процесс лиофилизации (давление, температуру, время) и обеспечить надежное воспроизведение приемлемого уровня влажности в готовом продукте, следует проводить анализ содержания влаги в готовом продукте и документально оформлять его результаты. Отклонения в поддержании требуемого уровня влажности в готовом продукте могут привести к тому, что готовый продукт будет менее активным и менее стабильным, чем это указано на этикетке.
21.4 Маршрут процесса
Планировочное решение помещения, относящееся к оборудованию наполнения, лиофилизации и укупоривания, должно быть выполнено так, чтобы перемещение открытого продукта было сведено до минимума.
21.5 Открытые контейнеры (первичная упаковка) и пробки
21.5.1 Упаковка подлежащего лиофилизации продукта остается открытой во время процесса сушки. В связи с этим продукт должен быть защищен от загрязнения во время перемещения от зоны наполнения в лиофильную установку, нахождения внутри морозильной камеры (сублиматора) и в конце процесса сушки до укупоривания.
21.5.2 Пробки, которые используются для лиофилизированного продукта, как правило, отличаются по конструкции от пробок, используемых в асептических процессах без лиофильной сушки, и пробок, используемых в процессах с финишной стерилизацией. Пробки с прорезью, применяемые при лиофилизации, могут потребовать специальных ограничительных элементов во время операции надевания, после которой пробка оказывается слегка надетой на флакон перед транспортированием в лиофильную установку. После завершения лиофилизации производится плотное укупоривание пробкой за счет давления противня или полки.
21.6 Перемещение в лиофильную установку
21.6.1 Перемещение наполненного продукта, принадлежностей и других предметов в лиофильную установку должно проводиться в критической зоне.
21.6.2 Должна быть разработана инструкция по загрузке лиофильной установки.
Дополнительные защитные меры могут включать в себя:
a) закрытые или полузакрытые передаточные кассеты;
b) камеры с однонаправленным потоком или камеры с избыточным давлением для перемещения кассет;
c) обеспечение однонаправленного потока над зоной загрузки.
Автоматические или механические системы передачи являются предпочтительными по сравнению с перемещением вручную.
21.6.3 Валидация перемещения продукта в лиофильную установку и из нее должна включать:
a) аттестацию помещений и укрытий с фильтрованным воздухом с использованием как физических, так и микробиологических методов;
b) аттестацию персонала с использованием микробиологических методов (см. 4.2);
c) использование имитаторов продукции или имитирующего бульона с применением существующих методов и условий передачи, исключая операцию лиофилизации.
21.7 Выгрузка и перемещение к оборудованию укупоривания
21.7.1 Должна быть разработана инструкция по разгрузке лиофильной установки.
21.7.2 Процесс лиофилизации и период времени между завершением цикла и выгрузкой после лиофилизации должны быть сокращены, насколько это возможно.
21.7.3 Сброс вакуума должен проводиться стерильным агентом, таким как воздух, азот или другой пригодный газ, который соответствует требованиям фармакопеи и прошел мембранную фильтрацию.
21.7.4 Перемещение открытого лиофилизированного продукта к оборудованию укупорки должно производиться в критической зоне.
Примечание - Если укупоривание флаконов выполняется внутри лиофильной установки, то перемещение может проводиться в менее критической зоне.
21.8 Очистка и дезинфекция лиофильной установки
Следует должным образом проводить очистку и дезинфекцию внутренних поверхностей лиофильной установки, включая линии стока водного конденсата.
21.9 Стерилизация лиофильной установки
21.9.1 Периодичность
21.9.1.1 Лиофильная установка должна проходить стерилизацию через определенные интервалы времени.
21.9.1.2 Эти интервалы, которые могут зависеть от вида продукта или других факторов, должны быть подтверждены при микробиологической валидации.
21.9.1.3 Должен быть определен максимально допустимый интервал времени между стерилизацией и началом цикла лиофильной сушки.
21.9.2 Методы
Методы стерилизации лиофильных установок должны быть валидированы.
Примечание - Методы стерилизации лиофильных установок паром или газом являются предпочтительными.
21.10 Система вентиляционных фильтров
21.10.1 При сбросе вакуума с сохранением герметичности (целостности) лиофильной установки должна использоваться система фильтрации газов. После каждого цикла лиофильной сушки следует проверять целостность фильтров. Следует избегать появления перепада давления на фильтре.
Примечание - Если применяются фиксированные газовые линии, должна быть предусмотрена система определения наличия газа.
21.10.2 Конструкция системы фильтрации должна допускать ее стерилизацию. По возможности, предпочтительно проводить контроль и стерилизацию "на месте", чтобы не прибегать к асептической замене фильтров.
21.11 Мониторинг окружающей среды по микроорганизмам и частицам
Должна быть введена программа микробиологического и физического контроля перемещения продукта и проведения лиофилизации (см. разделы 15-17).
21.12 Особенности наполнения средами для лиофилизации
21.12.1 Лиофилизированный продукт может представлять определенные трудности во время наполнения имитирующими средами (имитатором) из-за повышенного числа манипуляций и вмешательства персонала.
21.12.2 При валидации методов очистки, дезинфекции и стерилизации должно быть показано, что пролитые среды во время проведения работ с имитатором удаляются.
21.12.3 Испытания с наполнением средами должны имитировать условия окружающей среды и течение технологического процесса, включая: наполнение продуктом, предварительное надевание колпачков, загрузку и перемещение к лиофильной установке, выгрузку, заполнение лиофильной установки, процесс вакуумирования, плотное укупоривание пробкой за счет давления противня или полки.
21.12.4 В течение процесса могут производиться ручные манипуляции, включая удаление крышек противней или поддонов в камере при загрузке флаконов и ампул, выполнение соединений термопар или термопроб и разгрузку камеры. Во время выполнения регламентной работы по наполнению средами должны быть учтены все ручные манипуляции и вмешательства.
21.12.5 Наполнение средами должно дублировать процесс лиофилизации насколько это возможно, за исключением стадии замораживания, исходя из практических соображений, включая частичное вакуумирование и продолжительность, соответствующую среде.
21.12.6 Следует избегать фактического замораживания раствора.
Примечание - Если лиофилизация нерасфасованного продукта проводится не во флаконе или ампуле, то вмешательство человека в процесс лиофилизации увеличивается. В таких случаях лиофилизацию проходят целые противни с материалом. Материал должен измельчаться, чтобы продукт превратился в однородную пудру до продолжения дальнейшего асептического процесса. При этом продукт оказывается открытым окружающей среде в течение более длительного времени, что может повлиять на гарантию стерильности продукта.
21.13 Техническое обслуживание
21.13.1 В связи с тем, что при лиофилизации используются большие перепады температуры и давления, должна быть принята программа профилактического технического обслуживания
21.13.2 Следует проводить регулярные проверки, чтобы убедиться, что клапаны открываются и закрываются в нужное время и в нужном порядке.
ПРИЛОЖЕНИЕ А (справочное). Расчет риска контаминации для данного числа единиц, наполненных средами
ПРИЛОЖЕНИЕ А
(справочное)
Примечание - Приведенная информация является ознакомительной и представляет собой инструкцию, которая не носит обязательного характера.
Если число наполненных контейнеров ![]() относительно велико и вероятность контаминации
относительно велико и вероятность контаминации ![]() любого конкретного контейнера весьма мала, то число контаминированных единиц может быть распределено по закону Пуассона. Контрольное число единиц, в котором с 95%-ной вероятностью будет находиться хотя бы одна нестерильная единица
любого конкретного контейнера весьма мала, то число контаминированных единиц может быть распределено по закону Пуассона. Контрольное число единиц, в котором с 95%-ной вероятностью будет находиться хотя бы одна нестерильная единица ![]() , следуя закону Пуассона, приближенно можно вычислить следующим образом:
, следуя закону Пуассона, приближенно можно вычислить следующим образом:
![]() .
.
Это дает ![]() 2996 при
2996 при ![]() 0,001 (т.е. 0,1%). С другой стороны может быть использована более точная формула биномиального распределения:
0,001 (т.е. 0,1%). С другой стороны может быть использована более точная формула биномиального распределения:
![]() ,
,
где ![]() - доля нестерильных единиц. Это дает
- доля нестерильных единиц. Это дает ![]() 2995 при
2995 при ![]() 0,95.
0,95.
При наблюдаемом числе нестерильных единиц с 95%-ной вероятностью можно утверждать, что истинное число нестерильных единиц будет равно или менее верхнего 95%-ного доверительного предела. Следовательно, верхний доверительный предел должен использоваться для расчета "наихудшего случая" применительно к риску контаминации с помощью наблюдаемого числа нестерильных единиц.
Используя таблицу А.1, можно рассчитать риск контаминации для данного количества наполненных единиц.
Таблица А.1 - 95%-ный верхний доверительный предел для распределения Пуассона
| Полученная частота появления нестерильных единиц при наполнении средами | 95%-ный верхний доверительный предел |
| 0 | 2,9957 |
| 1 | 4,7439 |
| 2 | 6,2958 |
| 3 | 7,7537 |
| 4 | 9,1537 |
| 5 | 10,5130 |
| 6 | 11,8424 |
| 7 | 13,1481 |
| 8 | 14,4346 |
| 9 | 15,7052 |
| 10 | 16,9622 |
| 11 | 18,2075 |
Пример. Если из 3000 наполненных единиц одна оказалась нестерильной, то верхний 95%-ный доверительный предел риска контаминации будет не более чем 4,74/3000х100% =0,15%. Это не может считаться приемлемым.
Если из 10000 наполненных единиц три оказались нестерильными, то верхний доверительный предел риска контаминации будет не более чем 7,75/10000х100% =0,0775%.
В таблице А.2 содержатся результаты применения приведенных расчетов для различных по объему единиц, наполненных средами, и различного (увеличивающегося) количества нестерильных единиц. Для этого примера уровень тревоги принят равным >0,05% (доля контаминированных единиц). Уровень действия равен 0,10%. Предложенный уровень тревоги располагается внутри области, ограниченной жирными линиями таблицы. Уровень действия расположен выше и справа от жирной линии.
Например, если из 4750 наполненных единиц обнаружена одна контаминированная единица, то риск контаминации равен 0,0998%. Две контаминированных единицы из того же количества наполненных единиц дают риск контаминации 0,1325% и поэтому превышают уровень действия.
Все случаи микробной контаминации должны быть расследованы для установления причин, несмотря на данные, представленные в таблице А.2.
Таблица А.2 - Риск загрязнения (%) при 95%-ном доверительном интервале для возрастающего числа контаминированных единиц в одном прогоне наполнения средами
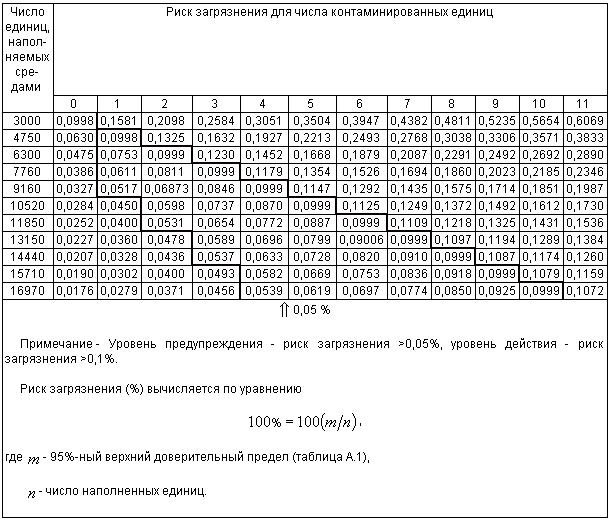
ПРИЛОЖЕНИЕ В (справочное). Библиография
ПРИЛОЖЕНИЕ В
(справочное)
[1] ГОСТ Р ИСО 11137-2000 Стерилизация медицинской продукции. Требования к валидации и текущему контролю. Радиационная стерилизация
[2] ГОСТ Р ИСО 11134-2000 Стерилизация медицинской продукции. Требования к валидации и текущему контролю. Промышленная стерилизация влажным теплом
[3] ИСО Р 14160-98 Стерилизация одноразовых медицинских изделий, содержащих материалы животного происхождения. Валидация и текущий контроль стерилизации с помощью жидких стерилизующих средств - ISO 14160:1998 - Sterilization of single use medical device, incorporating materials of animal origin - Validation and routine control of sterilization by liquid sterilants
[4] ГОСТ Р ИСО 11135-2000 Медицинские изделия. Валидация и текущий контроль при стерилизации оксидом этилена
[5] ГОСТ Р ИСО 9001-96 Системы качества. Модель обеспечения качества при проектировании, разработке, производстве, монтаже и обслуживании
[6] ГОСТ Р ИСО 9002-96 Системы качества. Модель обеспечения качества при производстве, монтаже и обслуживании